Traducción para Solicitudes a la EMA: Requisitos Regulatorios Farmacéuticos en la UE
La traducción para solicitudes a la EMA exige precisión, conocimiento regulatorio y cumplimiento riguroso de las plantillas Quality Review of Documents (QRD) de la Agencia Europea de Medicamentos. Las empresas farmacéuticas que buscan autorización de comercialización a través del Procedimiento Centralizado deben traducir la información del producto a todas las lenguas oficiales de la UE dentro de plazos exigentes — frecuentemente solo cinco días tras el dictamen positivo. Esta guía explica el marco regulatorio, los requisitos documentales y los estándares de calidad que definen solicitudes exitosas a la EMA.
Índice
Comprender los Requisitos de Traducción para Solicitudes a la EMA
La Agencia Europea de Medicamentos funciona como la puerta de entrada regulatoria para productos farmacéuticos que acceden al mercado europeo a través del Procedimiento Centralizado. Esta vía permite a las empresas presentar una única solicitud de autorización de comercialización que, una vez aprobada, otorga acceso simultáneo a todos los Estados miembros de la UE.
La traducción para solicitudes a la EMA implica la conversión de documentación regulatoria del inglés — la lengua de trabajo de la Agencia — a las 24 lenguas oficiales de la UE, más el noruego e islandés. El alcance de la traducción va más allá de la simple conversión lingüística; exige conocimiento regulatorio, dominio del área terapéutica y cumplimiento riguroso de las plantillas QRD de la EMA.
El Contexto Regulatorio de la Traducción para Solicitudes a la EMA
El Comité de Medicamentos de Uso Humano (CHMP) evalúa las solicitudes de autorización de comercialización y emite recomendaciones a la Comisión Europea. Tras un dictamen positivo del CHMP, los titulares de autorización de comercialización disponen de una ventana comprimida para completar todas las traducciones necesarias. Este calendario genera una presión significativa sobre las empresas farmacéuticas y sus socios de servicios de traducción.
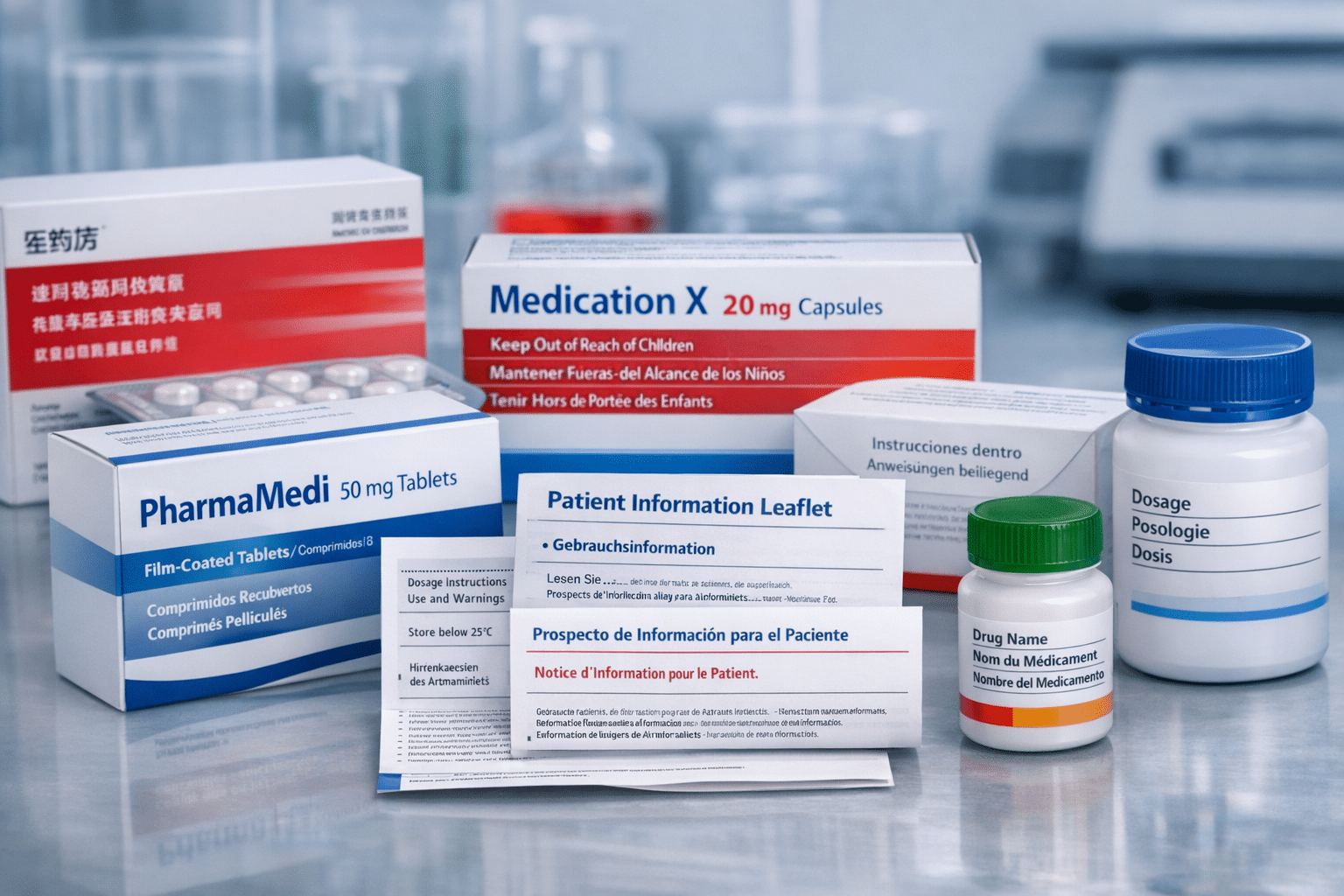
El marco regulatorio determina que la información del producto — comprendiendo la Ficha Técnica (FT), el prospecto y el etiquetado — esté disponible en todas las lenguas oficiales antes de que la Comisión Europea pueda conceder la autorización de comercialización. La calidad de la traducción tiene impacto directo en la seguridad de los pacientes y en la conformidad regulatoria. Para empresas españolas familiarizadas con los procedimientos de la AEMPS, el Procedimiento Centralizado representa una vía complementaria para productos específicos.
Documentos Clave que Requieren Traducción
Las solicitudes a la EMA abarcan varias categorías de documentos, cada una con requisitos específicos de traducción y estándares de calidad.
La Ficha Técnica (FT) o Resumen de Características del Producto proporciona a los profesionales sanitarios información esencial para la prescripción. Este documento abarca indicaciones, posología, contraindicaciones, advertencias, interacciones y propiedades farmacológicas. La traducción de la FT requiere especialistas en traducción médica que comprendan tanto la terminología clínica como las convenciones regulatorias.
El prospecto comunica directamente con los pacientes y debe utilizar lenguaje accesible manteniendo la precisión médica. Los traductores deben equilibrar la precisión técnica con la legibilidad, garantizando que los pacientes puedan comprender las instrucciones de dosificación, los potenciales efectos adversos y la información de seguridad.
El etiquetado abarca el embalaje exterior, el acondicionamiento primario y cualquier material acompañante. Frecuentemente se aplican requisitos específicos por país, particularmente para los contactos de notificación de reacciones adversas a medicamentos, que varían por Estado miembro.
El Proceso de Revisión Lingüística
La Revisión Lingüística de la EMA representa un punto de control de calidad crítico en el proceso de autorización de comercialización. Este procedimiento estandarizado de 25 días ocurre tras el dictamen e implica una revisión sistemática por las autoridades nacionales competentes en todos los Estados miembros.
Tras el dictamen del CHMP (Día 0), la información del producto anotada en inglés desencadena la fase de traducción. Hasta el Día 5, los titulares de autorización de comercialización deben enviar las versiones traducidas a los Estados miembros a través del sistema Eudralink. Las autoridades nacionales proceden entonces a la revisión lingüística durante 14 días naturales, devolviendo los ficheros revisados hasta el Día 19.
El titular de autorización de comercialización debe incorporar los comentarios de la revisión y enviar las traducciones finales hasta el Día 25. Este calendario comprimido deja margen mínimo para errores, haciendo esencial la preparación anticipada. Las empresas que inician la traducción antes del dictamen final — típicamente alrededor del Día 180 — se posicionan para el éxito.
Conformidad con la Plantilla QRD
El Grupo de Trabajo Quality Review of Documents desarrolla y mantiene plantillas estandarizadas que rigen el formato de la información del producto en toda la UE. La conformidad con la plantilla QRD es obligatoria, no opcional.
Las plantillas especifican encabezados obligatorios, declaraciones estándar, convenciones de formato y traducciones preaprobadas para términos comunes. La versión actual (10.4) aborda requisitos específicos para diferentes tipos de productos, incluyendo medicamentos de terapia avanzada y biosimilares.
Los traductores que trabajan en solicitudes a la EMA deben tener acceso directo a las plantillas QRD y documentos de referencia. Glosarios personalizados construidos a partir de terminología aprobada por el QRD garantizan consistencia en todos los conjuntos de documentos y pares lingüísticos.
Los elementos clave de conformidad incluyen especificaciones de fuente tipográfica, estructuras de encabezados, requisitos del triángulo negro para monitorización adicional e información de contacto obligatoria para notificación de farmacovigilancia en cada Estado miembro.
Estándares de Calidad para Traducciones EMA
La traducción regulatoria exige procesos de garantía de calidad que exceden los requisitos de la traducción comercial estándar. La certificación ISO 17100 proporciona un marco internacionalmente reconocido para la calidad de los servicios de traducción, exigiendo traductores cualificados, revisión por segundos lingüistas y sistemas documentados de gestión de la calidad.
Para solicitudes a la EMA, la calidad se extiende más allá de la precisión lingüística, abarcando la conformidad regulatoria, la adecuación terapéutica y la adherencia al formato. Los procesos de control de calidad deben verificar que las traducciones corresponden a las especificaciones de la plantilla QRD, utilizan terminología aprobada de forma consistente y mantienen la precisión de las referencias cruzadas entre FT, prospecto y etiquetado.
El proceso de revisión exhaustiva para traducciones farmacéuticas incluye típicamente traducción por expertos en la materia, revisión por revisores independientes con experiencia regulatoria, verificación terminológica contra glosarios aprobados, verificación de formato contra plantillas QRD y revisión final antes del envío.
Gestionar Plazos Ajustados
La ventana de cinco días post-dictamen representa uno de los plazos más desafiantes en la traducción farmacéutica. Una gestión exitosa requiere planificación estratégica que comienza meses antes del dictamen esperado del CHMP.
Las empresas proactivas establecen colaboraciones de traducción temprano en el proceso regulatorio. Proporcionan a los traductores versiones evolutivas de los documentos, permitiendo el desarrollo terminológico y la alineación de estilo antes de que las traducciones finales se vuelvan urgentes. Este enfoque permite que los traductores nativos se familiaricen con la terminología específica del producto y desarrollen glosarios personalizados apropiados.
Los procesos de revisión interna deben completarse antes del Día 210. Los titulares de autorización de comercialización que intentan revisar traducciones durante los cinco días finales arriesgan perder el plazo de envío o comprometer la calidad.
La tecnología de memoria de traducción acumula segmentos aprobados a lo largo de las iteraciones de los documentos, reduciendo el tiempo de entrega para actualizaciones mientras mantiene la consistencia. Esto resulta particularmente valioso para variaciones e informes periódicos de actualización de seguridad (PSUR) que modifican información del producto existente.
Seleccionar un Socio de Traducción
La traducción para solicitudes a la EMA requiere conocimientos especializados que van más allá de las capacidades generales de traducción farmacéutica. Para empresas españolas del sector farmacéutico y biotecnológico, la selección de un socio con experiencia demostrada es fundamental.
Evalúe potenciales socios por su experiencia en traducción regulatoria, particularmente con solicitudes del procedimiento centralizado. Solicite casos de estudio que demuestren la finalización exitosa de traducciones post-dictamen dentro de los plazos de la EMA. Verifique la certificación ISO 17100 e infórmese sobre la especialización en áreas terapéuticas relevantes para sus productos.
La capacidad representa una consideración crítica. Las solicitudes a la EMA requieren traducción simultánea a 24+ lenguas, cada una exigiendo traductores y revisores cualificados con experiencia farmacéutica. Los socios deben demostrar la profundidad de su red y planificación de contingencia para proyectos urgentes.
La infraestructura tecnológica es igualmente importante. Sistemas de memoria de traducción, plataformas de gestión terminológica y capacidades de transferencia segura de ficheros agilizan los flujos de trabajo y protegen información regulatoria confidencial. La integración con sus sistemas internos puede acelerar aún más la colaboración.
Considere el valor de una colaboración a largo plazo en lugar de relaciones transaccionales. Socios que comprenden sus productos, áreas terapéuticas e historial regulatorio entregan resultados más consistentes y se adaptan más eficientemente a requisitos urgentes. Consulte los testimonios de clientes para evaluar la fiabilidad y calidad del servicio.
¿Listo para discutir sus requisitos de traducción para solicitudes a la EMA? Solicite un presupuesto para una propuesta detallada adaptada a su calendario regulatorio y necesidades de documentación.